The name “orphan” can seldom be spoken without a sad tone, and never was this more true than in the case of orphan drugs. The terminology originates from orphan diseases, referring to diseases for which no curative therapy or effective drug was available. Neither hospitals nor pharmaceutical manufacturers were able to provide solutions for these patient groups, not least because therapeutic development had proven scientifically difficult, commercially unattractive due to perceived low patient numbers, or because the underlying causality of the disease could not be elucidated or translated into a viable therapeutic strategy.¹
Historically, these patients’ stories were confined to sections of medical textbooks devoted to “untreatable diseases.” The prevailing mindset for physicians was to alleviate suffering where possible, but to abandon any realistic hope of curative intervention. Much like Rémi, the orphaned protagonist in Hector Malot’s 19th-century novel Sans famille (Alone in the World), patients with rare diseases often felt unwanted by healthcare systems and were forced to journey from clinic to clinic in search of answers.² Although many rare diseases still have an unknown cause, this paradigm has changed dramatically in recent decades as advances in molecular biology and genetics have enabled the identification of causal mechanisms and the development of therapies targeting fundamental disease drivers.³
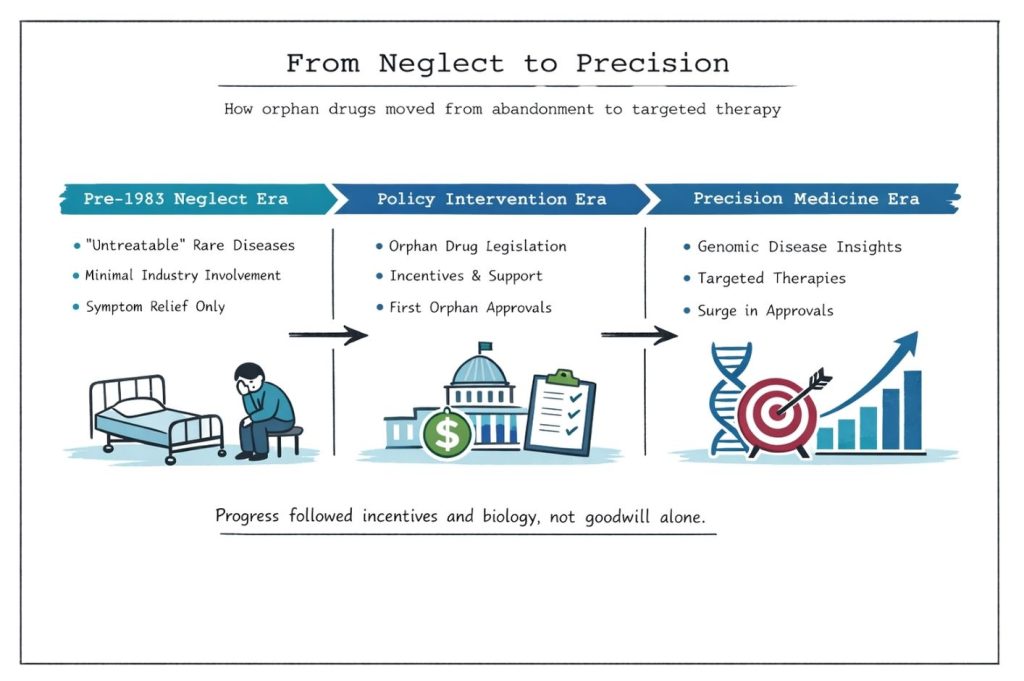
It is estimated that there are between 7 000 and 8 000 rare diseases, the majority of which are genetic in origin, yet for many the causative mutation or biological pathway remains unknown.⁴ Despite this, over the past 20 years the number of medicines developed for rare diseases has increased exponentially. In the United States alone, more than 1 100 orphan-designated drugs have now received approval.⁵ This growth did not occur spontaneously. A pivotal turning point was the introduction of the Orphan Drug Act (ODA) of 1983, which fundamentally altered the incentives governing drug development for small patient populations.
The ODA created a series of regulatory and financial incentives—including tax credits for clinical research, waiver of regulatory fees, protocol assistance, and seven years of market exclusivity—to encourage pharmaceutical companies to invest in rare disease therapeutics.⁶ As a result, development activity in orphan indications increased sharply. Other regions, including the European Union, Japan, and Australia, subsequently implemented comparable legislative frameworks. The first orphan drugs reached the market as early as 1985, followed by a steady and later near-exponential rise in approvals over subsequent decades.⁷
While the Orphan Drug Act repaired a failing economic mechanism by making development for small populations commercially viable, it was not the sole driver of innovation in this field. The completion of the Human Genome Project in 2003 marked a second transformative milestone.⁸ By enabling precise molecular and genetic characterization of disease, it allowed rare conditions to be defined with unprecedented specificity. This reduced diagnostic ambiguity and minimized inappropriate patient clustering, making it possible to target therapies to narrowly defined genetic subgroups. In contrast to the early post-ODA period—when treatments were largely developed for diseases with already well-understood mechanisms—the genomic era enabled therapeutic development for conditions that had previously been biologically opaque.³,⁹
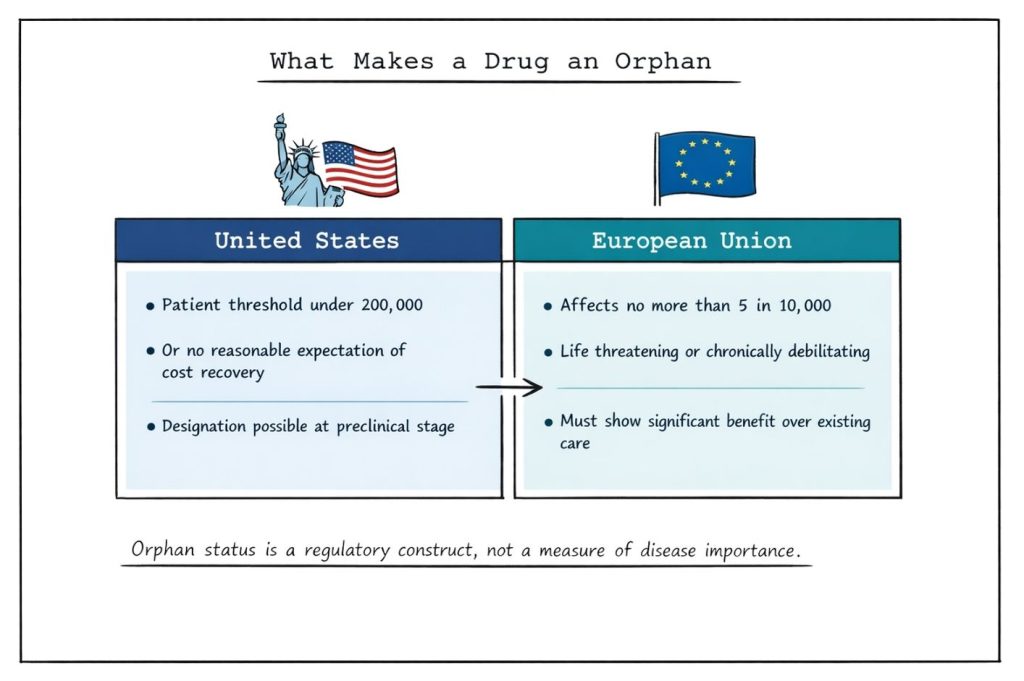
For a medicinal product to receive orphan designation, specific regulatory criteria must be met. In the United States, a disease must affect fewer than 200 000 individuals, or alternatively be one for which there is no reasonable expectation that development and marketing costs can be recovered from sales.¹⁰ In the European Union, the condition must be life-threatening or chronically debilitating, affect no more than 5 in 10 000 people, and either lack satisfactory treatment options or demonstrate significant clinical benefit over existing therapies.¹¹ These criteria are evaluated prior to marketing authorization and are distinct from requirements for approval.
Importantly, orphan designation may be granted at any stage of development, including preclinical phases, provided that sufficient scientific rationale exists. However, designation does not guarantee success. Many orphan-designated drugs require years of additional development and some ultimately fail to achieve marketing authorization due to lack of efficacy or unacceptable safety outcomes in pivotal trials. The withdrawal of previously approved orphan drugs following negative confirmatory studies underscores that regulatory incentives do not lower evidentiary standards for benefit-risk assessment.¹²
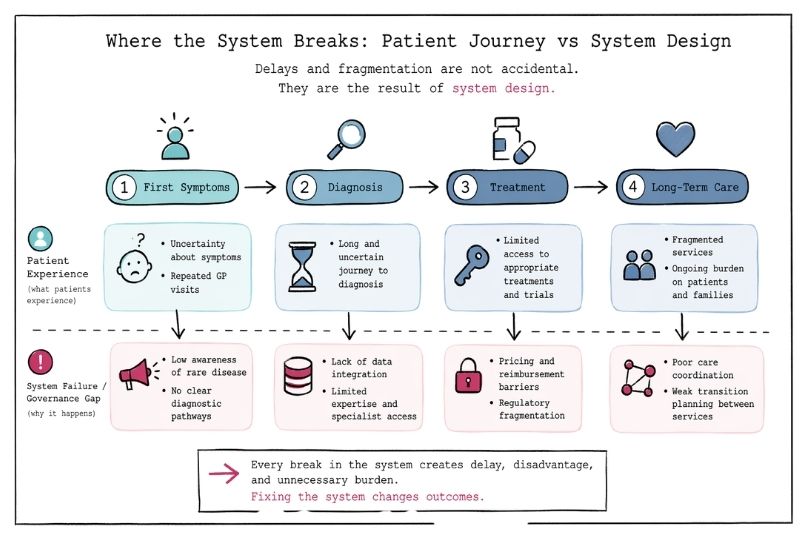
Clinical trial design in rare diseases poses unique challenges. Small patient populations limit statistical power, while phenotypic heterogeneity and poorly characterized natural histories further complicate interpretation. Consequently, traditional randomized controlled trials are often infeasible, necessitating innovative approaches such as adaptive designs, Bayesian statistics, use of external or historical controls, and reliance on surrogate or composite endpoints.¹³ Regulatory agencies increasingly accept such methodologies, provided they are scientifically justified and transparently reported, but inadequate trial design remains a major reason for failure in orphan drug development.¹⁴
Even after regulatory approval, market access and reimbursement remain significant hurdles. The value of orphan drugs is increasingly assessed not only through conventional cost-effectiveness metrics but also through broader considerations such as disease burden, severity, lack of alternatives, and societal costs avoided, including reduced hospitalizations, long-term disability, and caregiver burden.¹⁵ Health technology assessment frameworks vary widely between countries, often resulting in delayed or restricted access despite demonstrated clinical benefit.
In conclusion, although orphan drug legislation and genomic science have transformed the therapeutic landscape for rare diseases, the path from scientific discovery to patient access remains complex and uncertain. Regulatory incentives alone are insufficient to ensure success. Robust trial design, early stakeholder engagement, collaboration with patient organizations, and alignment between regulators, payers, and developers are essential to translate scientific advances into meaningful and sustainable outcomes for patients who have historically been overlooked.