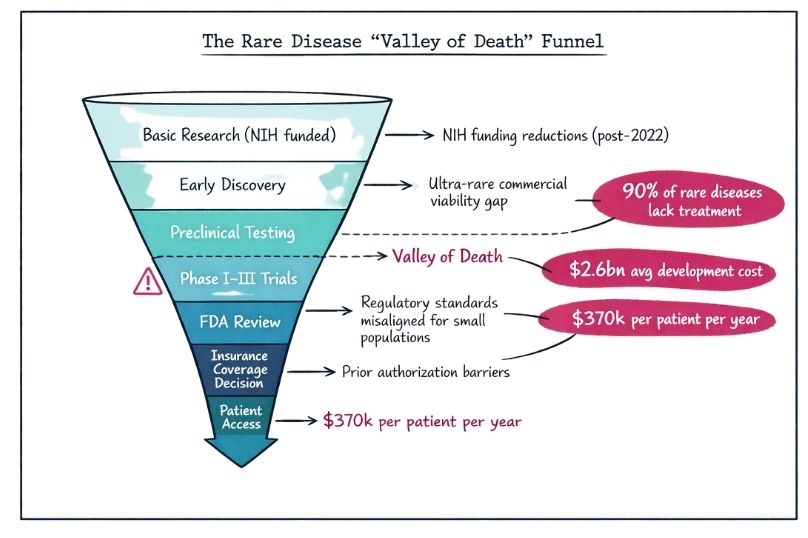
The landscape of rare disease treatment development has undergone a dramatic transformation in 2025, with the FDA implementing groundbreaking regulatory pathways that address the unique challenges faced by patients with ultra-rare conditions. These initiatives represent the most significant regulatory evolution in rare disease therapeutics in over a decade, offering new hope for the 30 million Americans living with one of the estimated 7,000 known rare diseases.
Traditional clinical trial methodologies have long posed insurmountable barriers for rare disease drug development, where patient populations may number in the hundreds or even fewer. The FDA's response has been comprehensive and innovative, introducing multiple complementary pathways designed to accelerate access to life-saving treatments while maintaining rigorous safety standards.
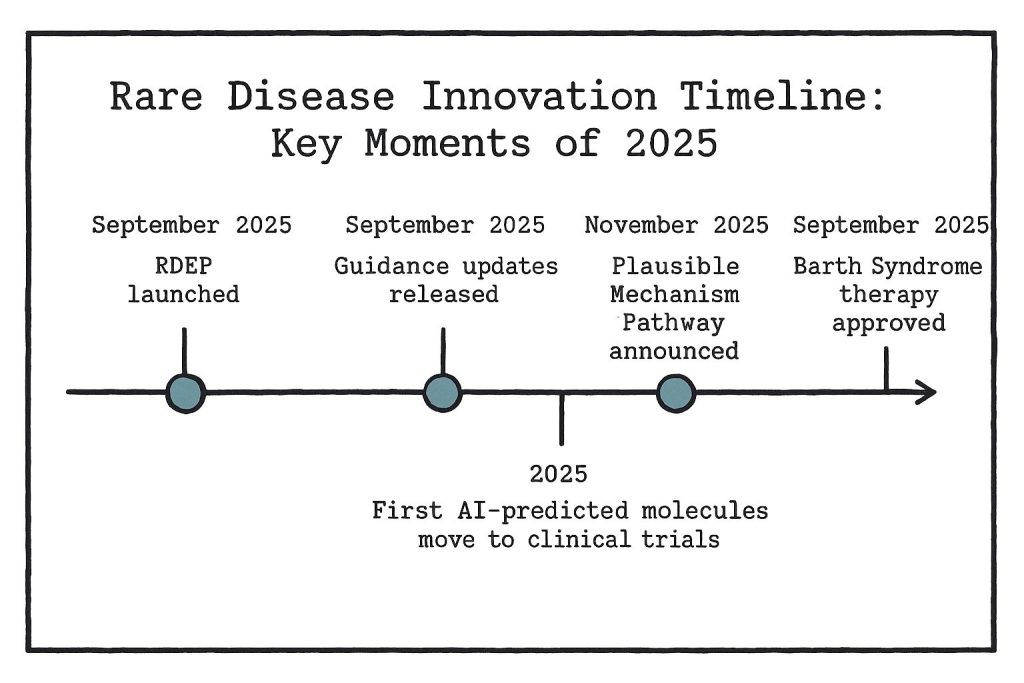
On November 12, 2025, FDA Commissioner Marty Makary and Center for Biologics Evaluation and Research (CBER) Director Vinay Prasad unveiled the most significant regulatory innovation in rare disease treatment approval: the Plausible Mechanism Pathway. This framework fundamentally reimagines how the agency evaluates therapies for conditions where randomized controlled trials are not feasible.
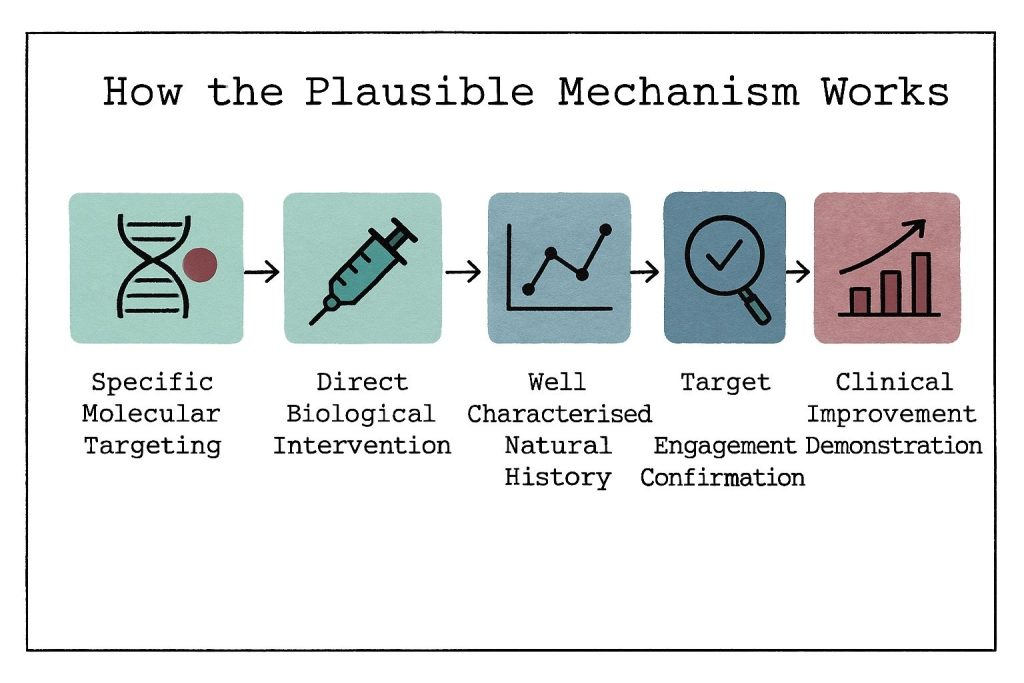
The pathway represents a paradigm shift from traditional evidence requirements, focusing instead on biological plausibility and mechanism-based approval criteria. This approach is particularly revolutionary for bespoke therapies addressing fatal or severely disabling childhood diseases, where waiting for conventional trial data could mean the difference between life and death for affected patients.
The Plausible Mechanism Pathway operates on five essential criteria that create a structured roadmap for product approval:
Specific Molecular Targeting: The pathway requires identification of a precise molecular or cellular abnormality, moving beyond broad diagnostic criteria to focus on the underlying biological mechanisms driving disease pathology.
Direct Biological Intervention: Medical products must demonstrate that they directly target the identified biological alterations, whether through gene editing, protein replacement, or other mechanism-based approaches.
Well-Characterized Natural History: Sponsors must provide comprehensive documentation of disease progression in untreated populations, establishing clear baseline expectations for clinical outcomes.
Target Engagement Confirmation: Evidence must confirm successful biological intervention, whether through gene editing verification, protein level restoration, or other measurable biological endpoints.
Clinical Improvement Demonstration: The pathway requires observable improvements in clinical outcomes or disease trajectory, though these may be measured through surrogate endpoints rather than traditional clinical measures.
While initially focused on cell and gene therapies, the FDA has explicitly stated that the pathway may expand to include small molecules and other therapeutic modalities, potentially revolutionizing treatment development across the rare disease spectrum.
Complementing the Plausible Mechanism Pathway, the FDA introduced the Rare Disease Evidence Principles (RDEP) in September 2025. This joint initiative between the Center for Drug Evaluation and Research (CDER) and CBER specifically addresses conditions with known genetic defects, extremely small patient populations (typically fewer than 1,000 patients in the United States), and significant unmet medical need.
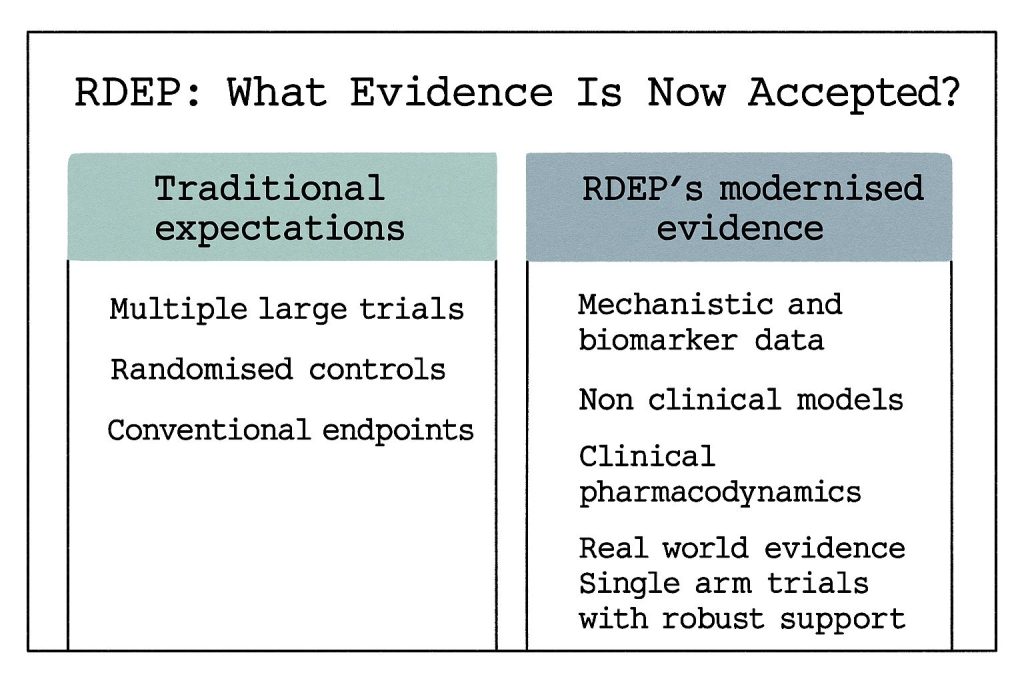
RDEP provides unprecedented clarity on acceptable evidence types, fundamentally changing sponsor expectations for regulatory submission requirements. The principles recognize that traditional Phase III trial designs are often impossible or unethical for ultra-rare conditions, requiring innovative approaches to evidence generation.
Under RDEP guidelines, the FDA now accepts diverse forms of evidence that were previously considered insufficient for drug approval:
Mechanistic and Biomarker Evidence: Strong biological rationale supporting therapeutic intervention, including biomarker data demonstrating target engagement and biological activity.
Non-Clinical Model Data: Evidence from relevant animal models, in vitro studies, and other pre-clinical research that demonstrates therapeutic potential and safety profiles.
Clinical Pharmacodynamic Data: Proof-of-concept studies showing biological activity and dose-response relationships in human subjects.
Real-World Evidence: Case reports, expanded access program data, and natural history studies that provide insights into treatment effects in clinical practice.
Perhaps most significantly, RDEP establishes that substantial evidence of effectiveness can be demonstrated through one adequate and well-controlled single-arm trial when accompanied by robust confirmatory data. This represents a fundamental departure from traditional two-trial requirements, acknowledging the practical impossibilities of conducting multiple large-scale trials in ultra-rare disease populations.
The FDA's commitment to rare disease innovation extends beyond pathway creation to comprehensive guidance updates released in September 2025. These three draft guidance documents provide detailed frameworks for sponsors navigating the complex landscape of rare disease therapeutic development.
The guidance on Innovative Designs for Clinical Trials of Cellular and Gene Therapy Products in Small Populations addresses critical methodological challenges facing rare disease researchers. The document provides specific recommendations for adaptive trial designs that can modify protocols based on accumulating data, novel endpoints that capture meaningful clinical benefits, and natural history comparators that eliminate the ethical concerns of placebo-controlled trials in fatal conditions.
The guidance emphasizes collaborative multi-site models that enable data sharing and protocol alignment across research centers, maximizing the utility of limited patient populations while maintaining scientific rigor.
Recognizing the unique challenges of monitoring rare disease treatments after approval, the FDA's guidance on Postapproval Methods to Capture Safety and Efficacy Data establishes frameworks for long-term surveillance in small populations. Traditional pharmacovigilance systems often fail to capture meaningful safety signals in rare disease populations due to limited patient numbers and diverse clinical presentations.
The new guidance outlines innovative approaches including patient registries, specialized surveillance networks, and real-world evidence collection methods that can provide meaningful safety and efficacy data over extended time periods.
The FDA's third guidance document updates Expedited Programs for Regenerative Medicine Therapies, expanding existing flexibility mechanisms while providing more detailed eligibility criteria and updated examples. These enhancements build upon established pathways including Breakthrough Therapy designation and Accelerated Approval, ensuring rare disease treatments can leverage multiple complementary regulatory mechanisms.
Breakthrough Therapy designation continues to provide intensive FDA guidance throughout development phases, enabling priority review and potential early approval for treatments demonstrating substantial improvement over existing therapeutic options. For rare diseases, where any effective treatment may represent a dramatic advancement, this designation has proven particularly valuable.
The Accelerated Approval Pathway remains crucial for ultra-rare diseases, allowing approval based on surrogate endpoints rather than traditional clinical outcomes. This mechanism has enabled numerous rare disease treatments to reach patients years earlier than would be possible under conventional approval timelines.
These regulatory innovations are already producing tangible results for rare disease patients. In September 2025, the FDA granted accelerated approval to Stealth BioTherapeutics' therapy for Barth Syndrome, an ultra-rare mitochondrial disorder affecting primarily young males. This approval followed a decade of patient-driven research and advocacy, demonstrating how new regulatory frameworks can translate years of scientific effort into accessible treatments.
The integration of artificial intelligence into rare disease drug discovery has further accelerated development timelines. AI-predicted molecules are successfully advancing into clinical trials for conditions including neurofibromatosis type 1, reducing early-stage development time from years to months in some cases.
Patient advocacy organizations have played increasingly important roles in these developments, providing natural history data, facilitating research participation, and advocating for regulatory flexibility. The FDA's enhanced engagement with patient communities reflects recognition that traditional regulatory approaches must adapt to the unique needs and constraints of rare disease research.
The FDA's 2025 regulatory innovations represent more than procedural updates; they signal a fundamental philosophical shift toward patient-centered drug development that prioritizes biological understanding over statistical power. This approach acknowledges that rare diseases require fundamentally different regulatory frameworks than common conditions, where large patient populations enable traditional clinical trial designs.
Looking forward, these pathways are expected to accelerate development timelines for hundreds of rare disease treatments currently in various stages of research and development. The combination of mechanism-based approval criteria, flexible evidence requirements, and enhanced regulatory guidance creates an environment where innovative therapies can reach patients more rapidly while maintaining appropriate safety oversight.
The success of these initiatives will likely influence regulatory approaches globally, as international agencies observe the FDA's experience with mechanism-based approvals and flexible evidence standards. This could lead to harmonized international standards that further accelerate rare disease treatment development worldwide.
For patients and families affected by rare diseases, these regulatory changes represent hope for conditions that have historically been overlooked by pharmaceutical development. The FDA's commitment to innovation while maintaining safety standards creates a framework where scientific advancement can translate more rapidly into clinical benefits, potentially transforming outcomes for millions of patients worldwide.