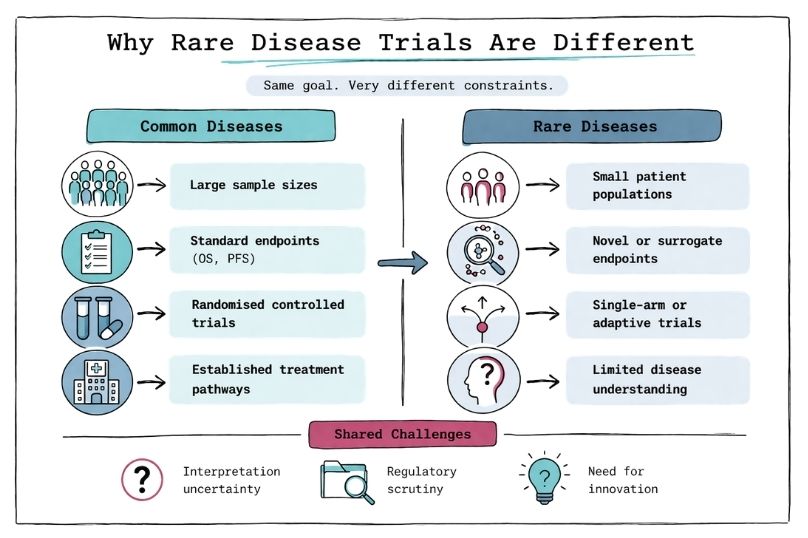
In rare diseases, as in more common conditions, regulatory approval requires clear evidence of clinical benefit from a therapy, as defined by agencies such as the European Medicines Agency and the U.S. Food and Drug Administration [1,2]. To demonstrate such benefit, clinical trials in rare diseases rely heavily on endpoints that can unambiguously show whether one therapy outperforms another, for example improvement in forced vital capacity (FVC) in idiopathic pulmonary fibrosis trials with nintedanib [3] or reduction in spleen volume in Gaucher disease [4].
Such as improved survival, reduced symptom burden, or better functional outcomes, for example increased six-minute walk distance in pulmonary arterial hypertension [5] or reduced frequency of vaso-occlusive crises in sickle cell disease with crizanlizumab [6]. Demonstrating such benefit through robust and reproducible evidence is the cornerstone of clinical development. However, statistical power is strongly influenced by sample size, and in rare diseases patient numbers are inherently limited, as seen in ultra-rare conditions such as Niemann–Pick disease type C [7]. This leads to the first major challenge: identifying and recruiting patients requires significant effort, including natural history studies and patient registries [8].
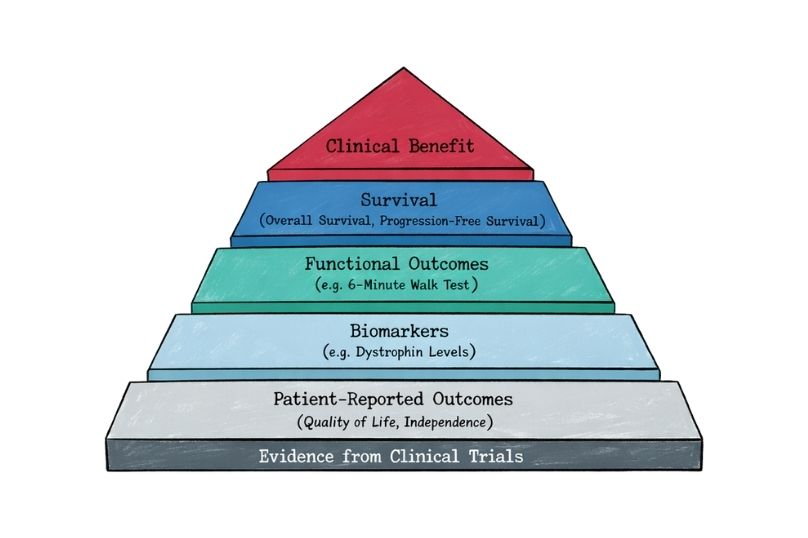
Traditional endpoints such as overall survival (OS) or progression-free survival (PFS), commonly used in oncology (e.g., pembrolizumab trials within the KEYNOTE clinical trial program), are often not feasible or informative in rare diseases due to slow disease progression, heterogeneity, or limited understanding of disease biology [9,10]. In many rare diseases, there is also no established standard-of-care comparator, as seen historically in spinal muscular atrophy prior to disease-modifying therapies such as nusinersen [11]. As a result, clinical trials may rely on external controls, historical cohorts, or consensus-defined standards of care to contextualize treatment effects [12].
These may include composite endpoints, functional scales, or biomarker-based surrogates, such as the NPC Clinical Severity Scale in Niemann–Pick disease type C [13] or dystrophin expression levels in Duchenne muscular dystrophy trials [14]. In oncology rare diseases such as neuroblastoma, endpoint development has also required adaptation, with composite measures such as event-free survival (EFS), minimal residual disease (MRD), and imaging-based response criteria (e.g., MIBG scoring systems) being used to better capture clinically meaningful outcomes in heterogeneous and aggressive disease settings [19,20]. Increasingly, regulators accept such innovative endpoints when they are well-validated and clinically meaningful, as illustrated by the approval of eteplirsen based on surrogate biomarker data [15].
The need to develop novel endpoints, combined with statistical approaches adapted to very small patient populations, often results in datasets that are less intuitive and more difficult to interpret compared to traditional large-scale trials [8,9]. For example, Bayesian statistical methods or adaptive trial designs are frequently employed to maximise information from limited sample sizes, but these approaches may be less familiar to regulators and clinicians, potentially leading to increased scrutiny during regulatory review [10,12]. Furthermore, rare disease trials are often designed using external controls, single-arm studies, or surrogate endpoints, which differ from the conventional randomised controlled trial paradigm and may raise questions regarding robustness and generalisability of the findings [2,12].
In addition, many rare diseases are characterised by slow progression and high inter-patient variability, meaning that measurable treatment effects may appear modest over the duration of a clinical trial. Small changes in functional scales or biomarker levels—while statistically significant—may be perceived as clinically marginal, making it challenging to clearly demonstrate meaningful patient benefit [9,13]. As a result, both regulators and health technology assessment bodies may question the clinical relevance of observed effects, particularly when long-term outcomes such as survival or irreversible disease modification cannot be readily demonstrated within feasible study timelines [1,2].
A critical and increasingly recognised dimension in rare disease trials is the role of patients and their caregivers in defining what constitutes meaningful benefit. Given the heterogeneity, low prevalence, and often poorly characterised natural history of many rare diseases, patients are uniquely positioned to articulate which outcomes truly matter in daily life, such as preservation of independence, reduction in caregiver burden, or slowing of functional decline. A notable example is Duchenne muscular dystrophy, where patient advocacy groups played a pivotal role during the regulatory review of eteplirsen, contributing patient-reported perspectives that influenced the U.S. Food and Drug Administration’s decision to grant accelerated approval despite modest dystrophin biomarker data [15,16]. Similarly, in spinal muscular atrophy, caregiver-reported outcomes and functional measures such as motor milestone achievement were critical in demonstrating the value of nusinersen beyond traditional endpoints [11,17].
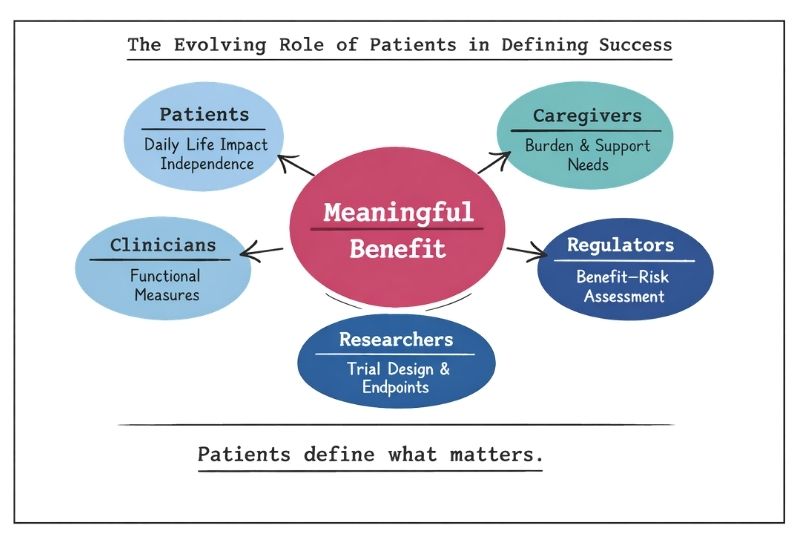
In contrast to more common diseases, where large datasets and well-established endpoints dominate decision-making, the patient perspective in rare diseases often carries proportionally greater weight in both regulatory and health technology assessment discussions. This is reflected in initiatives such as patient-focused drug development by the U.S. Food and Drug Administration and similar frameworks from the European Medicines Agency, which actively incorporate patient experience data into benefit–risk assessments [2,18]. In ultra-rare conditions, where quantitative endpoints may be limited or difficult to interpret, qualitative insights from patients and caregivers can be decisive in contextualising modest numerical improvements as clinically meaningful changes in quality of life. Consequently, integrating patient-reported outcomes and structured patient input is not only complementary but often essential in establishing the true value of therapies in rare disease settings.